Gregor Dückers und Tim Niehues, Krefeld
Primäre Immundefekte – Eine orientierende Übersicht
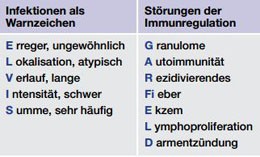
Tab. 1 Warnzeichen für primäre Immundefekte
Primäre Immundefekte treten meist durch eine pathologische Infektanfälligkeit in Erscheinung. Typische Warnsignale verbergen sich hinter den Akronymen ELVIS und GARFIELD (Tab. 1) (Farmand et al. 2011). PID können sich in allen Organen manifestieren, wobei häufig Haut, Lunge und Darm betroffen sind (Relan et al 2014; Jesenak et al. 2014; Uhlig et al. 2014). Nach einer detaillierten Anamneseerhebung und klinischen Untersuchung erfolgt das weitere diagnostische Vorgehen entsprechend der Vorgaben von Konsens- und evidenzbasierten Leitlinien (Farmand et al. 2011, de Vries 2011). In einzelnen komplexen Fällen, die mit bisherigen Methoden nicht aufgeklärt werden können, kann ein sogenanntes „Whole exome Sequencing“ indiziert sein (Platt 2014). Diagnostizierte PID lassen sich in eine der acht Hauptgruppen einordnen (Tab. 2) (Al-Herz 2014).
Kombinierte Immundefekte – Schwere kombinierte Immundefekte
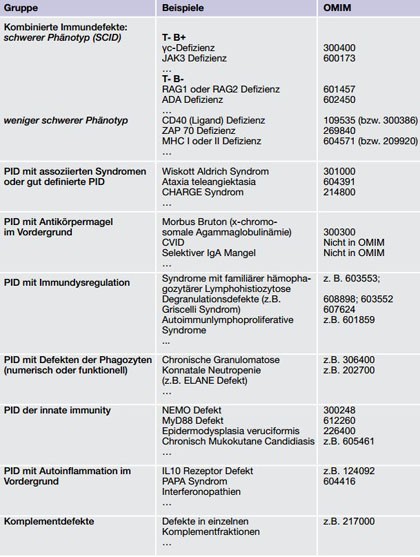
Tab. 2 (IUIS Klassifikation Primärer Immundefekte mit beispielhaften Erkrankungen, (adaptiert von Al Herz 2014): PID = Primäre Immundefekt, CVID = Common variable immunodeficiency, SCID, Severe Combined Immuno Deficiency)
Patienten, bei denen sowohl ein Antikörpermangel (Verminderung von Immunglobulinen) als auch eine Einschränkung der T-zellulären Funktion besteht, werden in die Gruppe der kombinierten Immundefekte klassifiziert. Diese Gruppe unterteilt sich in schwere kombinierte Immundefekte (SCID) und weniger schwere Formen. Bei einem SCID fehlen T- und B-Zellen vollständig oder die T-Zellen sind derart funktionsbehindert bzw. numerisch reduziert, dass keine reguläre B-zelluläre Antwort erfolgen kann. Die klassische SCID-Erkrankung manifestiert sich im Säuglingsalter und ist ohne Stammzelltransplantation tödlich (van der Burg & Gennery 2011). Weniger schwer ausgeprägte kombinierte PID können auch erst im Erwachsenenalter symptomatisch werden. In solchen Fällen werden gelegentlich hypomorphe Mutationen gefunden, die nur eine Restfunktion des betreffenden Proteins erhalten (z.B. RAG) (Felgentreff et al. 2011, Schütz 2008).
Typische
(opportunistische) Infektionen bei schweren kombinierten
Immundefekten im Säuglingsalter sind z.B. Pneumocystis jirovecii
oder schwere Virusinfektionen (CMV und EBV). Bei Erwachsenen mit in
der Regel weniger aus-
geprägtem
kombinierten Immundefekt sind typische Infektionen: Aspergillose,
Toxoplasmose, Adenovirus-Infektion oder auch
Pneumocystis-Infektionen, Kryptosporidiose, systemischer Candidose,
CMV Retinitis oder Kolitis, Mykobakteriose. Darüber hinaus haben die
Patienten gehäuft EBV-assoziierte Lymphome oder granulomatöse
Entzündungen (Rose et al. 2014). Letztere sind Ausdruck
unzureichender Erregerelimination und konsekutiver
autoinflammatorischer Lokalreaktion. Neben supportiven Maßnahmen
(z.B. IgG Substitution, PCP-Prophylaxe) gilt es, Infektionen früh,
aggressiv und gezielt antiinfektiös zu therapieren. Schließlich
stellt eine Stammzelltransplantation eine kurative Behandlungsoption
dar. Diese Möglichkeit ist im Säuglingsalter bei SCID
alternativlos, wohingegen für das Erwachsenenalter mit kombiniertem
PID nur wenige Erfahrungen vorliegen. Es besteht die Möglichkeit von
Screeninganalysen auf SCID im Neugeborenenalter.
PID mit assoziierten Syndromen oder gut definierte Immundefekte
In dieser PID Gruppe finden sich alle gut definierten PID mit nahezu pathognomischen, klinischem Phänotyp, z.B. Ataxia teleangiektasia (Ataxie + Teleangiektasien) oder Wiskott Aldrich Syndrom (Thrombozytopenie mit Mikrothrombozyten und Petechien). Eine Würdigung aller zugehörigen Entitäten würde den Rahmen dieser Übersicht sprengen. Daher verweisen wir auf entsprechende Übersichtsarbeiten, z.B. Kersseboom et al. (Kersseboom et al. 2011).
PID mit Antikörpermangel im Vordergrund
Den größten Anteil an PID haben Immundefekte mit Antikörpermangel im Vordergrund. Hierbei sind mindestens zwei Immunglobulinhauptklassen oder eine Haupt- und eine IgG-Subklasse vermindert. Beispielhaft für solche Immundefekte sind der variable humorale Immundefekt (= CVID, common variable immunodeficiency), die Agammaglobulinämie (Morbus Bruton), der selektive IgA-Mangel oder das Hyper-IgM-Syndrom (Driessen & van der Burg 2011). Im letzten Fall ist IgM erhöht oder normwertig und IgG sowie IgA vermindert. Der CVID ist der häufigste PID mit Antikörpermangel. Er führt zu rezidivierenden Infekten der Atemwege oft mit bekapselten Bakterien. Die Therapie von Patienten mit Antikörpermangel sollte der interdisziplinären AWMF Therapieleitlinie folgen (Krudewig et al. 2012). Sie beinhaltet im wesentlichen Infektionsprophylaxe und die Gabe von Immunglobulinen. Andernfalls birgt die chronische Erkrankung ein hohes Morbiditätsrisiko (z.B. Bronchiektasen oder Colitis).
PID mit Immundysregulation
Immundysregulation
kann ein maßgeblicher Hinweis auf Vorliegen eines PID sein. So
werden bei PID regelmäßig
assoziierte
Autoimmunphänomene, z.B. autoimmun vermittelten Zytopenien oder
Schilddrüsendysfunktionen beobachtet. Langsam progrediente
Lymphoproliferation, Splenomegalie oder Lymphknotenschwellung oder
akute Phasen von hämophagozytärer Lymphohistiozytose können so ein
Ausdruck angeborener Regulationsstörung sein. Beispiele für diese
Gruppe sind das Autoimmunlymphoproliferative Syndrom (ALPS), dem u.a.
ein Apoptosedefekt (Defekte des programmierten Zelltods) der
Lymphozyten oder Degranulationsdefekte (Defekte bei der Freisetzung
von zytotoxischen Effektormolekülen) wie z.B. das Griscelli Syndrom
zu Grunde liegen. Die Therapie umfasst in Abhängigkeit zur Klinik
abwartendes Verhalten bis hin zur Verabreichung einer starken
immunsuppressiven Therapie oder eine Stammzelltransplantation.
PID mit Defekten der Phagozyten
Bei PID mit Phagozytendefekten kann sowohl die Funktion gestört als auch die Anzahl der betreffenden Zellen vermindert sein. Angeborene Neutropenien, Leukozytenadhäsionsdefekte (LAD) und PID mit selektiver Infektanfälligkeit gegen Mykobakterien (MSDM, Mendelian susceptibility to mycobacterial disease) (Andrews & Sullivan 2003) zählen zu dieser Klasse. Funktionelle, intrazelluläre Enzymdefekte finden sich beispielhaft bei der chronischen Granulomatose (CGD) als einem Prototyp für diese Gruppe. Bei der CGD können wegen eines Enzymdefekts keine antibakteriellen, reaktiven Sauerstoffspezies gebildet werden. Das mangelhafte Abtöten der phagozytierten, d.h. intrazellulären Erreger führt zu invasiven bakteriellen, (pyogenen) Infektionen und Mykosen und schließlich zu Granulombildung (Seger 2010). Typische Erreger in Haut-, Lymphknoten- oder Leberabszessen sind Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens. Aspergillen können bei betroffenen CGD Patienten regelmäßig aus Lungen oder Knochen isoliert werden. Therapeutisch müssen betroffene Patienten eine Dauerprophylaxe mit z.B. Cotrimoxazol und Itraconazol erhalten. CGD kann mit einer Stammzelltransplantation geheilt werden.
PID der Innate Immunity
Patienten
mit Defekten im Bereich der innate immunity haben ebenfalls
wiederkehrende invasive Infektionen. Dies kann pyogene Bakterien,
Pneumokokken, Staphylokokken (MyD88-, IRAK4-Defekte) oder Warzen
(WHIM Syndrom, Epidermodysplasia veruciformis) oder auch
Pilzinfektionen betreffen (Chronisch Mukokutane Candidiasis).
Definierte
Defekte in der
innate immunity betreffen zelluläre Immunantworten nach
Zellaktivierung über sogenannte Tolllike Rezeptoren (TLRs) oder z.B.
innerhalb der Signalverarbeitung über den NF-κB Weg.
PID mit Autoinflammation im Vordergrund
In letzter Zeit werden zunehmend neue, monogenetische Autoinflammationserkrankungen aufgeklärt. Deren Phänotypen haben Überlappungen zu den bisher aufgeführten klassischen primären Immundefekten (Martinon et al. 2015; Dueckers et al. 2014). Leitsymptome von schweren Autoinflammationserkrankungen sind u. a. periodisches oder persistierendes Fieber unklarer Ursache, Polyserositis, Exanthem und auch schwere oder frühmanifeste (<1. Lebensjahr) Colitis. Breite immunsuppressive Therapie oder cytokinspezifische Inhibition der Autoinflammation kann indiziert sein.
Komplementdefekte
Einer
der entwicklungsgeschichtlich ältesten Bestandteile unseres
Immunsystems ist das Komplementsystem. Streng reguliert, ähnlich
einer Gerinnungskaskade können vor allem bekapselte Keime (z.B.
Meningokokken) u.a. durch Complementfaktoren perforiert und getötet
werden. Auch bei der Regulation von Immunantworten sowie Beseitigung
von Immunkomplexen ist das Komplement involviert. Hieraus erklärt
sich das begünstigte Auftreten eines Systemischen Lupus
Erythematodes oder anderen Autoimmunerkrankungen. Für alle Proteine
der Komplementkaskade C1 bis
C9
sind Defekte beschrieben (Grumach 2014; Degn 2011). Im
Lektin-Aktivierungsweg gibt es beschriebene Defizienzen u.a. des
Mannanbindenden Lektins (MBL) (Heitzeneder 2012). Defekte bestimmter
Regulatorproteine innerhalb des Komplementaktivierungsweges können
wieder zu ganz unterschiedlichen Erkrankungen führen, z.B.
hereditäres Angioödem, atypisch verlaufendes hämolytisches
urämisches Syndrom oder paroxysmale, nächtliche
Hämoglobinurie.
Da eine regelmäßige Substitution von Einzelfaktoren [C1-C9] derzeit
nicht möglich ist, kommt der Prävention große Bedeutung zu.
Impfungen gegen Meningokokken, HiB und Pneumokokken sind sinnvoll.
Fazit
Das klassische Spektrum von PID ist sehr variabel und erfordert eine interdisziplinäre Zusammenarbeit von allen in der Patientenversorgung involvierten Personen. Die Diagnostik reicht von gründlicher Anamneseerhebung bis zu hochspezialisierter, aufwändiger Labordiagnostik. Ebenso stellt sich das therapeutische Vorgehen komplex dar. Genügt bei einigen Patienten mit PID das Beachten von einfachen Hygienemaßnahmen und Einnahme einer präventiven Medikation oder Gabe von Immunglobulinen, müssen andere Patienten z.B. mit SCID umgehend einer Stammzelltransplantation zugeführt werden. Die Diagnostik und Therapie sollte möglichst in Zusammenarbeit mit einem Immundefektzentrum erfolgen. Kontakt kann über folgende Links hergestellt werden: www.dsai.de, www.find-id.net und www.immundefekt.de.
Abkürzungen
CGD, chronic granulomatous disease; CMV, Cytomegalovirus; CVID, common variable immunodeficiency; EBV, Epstein-Barr virus; Ig, Immunglobulin; MBL, Mannan-bindendes Lektin; MSDM, Mendelian susceptibility to mycobaterial disease; NK-Zellen, natürliche Killerzellen; RAG, Rekombinationsaktivierendes Gen; SCID, Schwerer kombinierter Immundefekt; SZT, Stammzelltransplantation
Literatur
RAl-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, Etzioni A, Franco JL, Gaspar HB, Holland SM, Klein C, Nonoyama S, Ochs HD, Oksenhendler E, Picard C, Puck JM, Sullivan K, Tang ML. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol.2014 Apr 22;5
Andrews T, Sullivan KE (2003) Infections in patients with inherited defects in phagocytic function. Clin Microbiol Rev 16:597-621
de Vries E; European Society for Immunodeficiencies (ESID) members (2012) Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol 167:108-119
Degn SE, Jensenius JC, Thiel S (2011) Disease-causing mutations in genes of the complement system. Am J Hum Genet 88:689-705
Driessen G, van der Burg M (2011) Educational paper: primary antibody deficiencies. Eur J Pediatr 170:693-702
Dueckers G, Sander O, Niehues T. (2014) Autoi flammatory Diseases. Klin Padiatr. 226(3):133-42
Farmand S, Baumann U, von Bernuth H, Borte M, Förster-Waldl E, Franke K, Habermehl P, Kapaun P, Klock G, Liese J, Marks R, Müller R, Nebe T, Niehues T, Schuster V, Warnatz K, Witte T, Ehl S, Schulze I (2011) AWMF Leitlinie „Diagnostik von primären Immundefekten“. AWMF: Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften (e.V.). www.awmf.org/leitlinien/detail/ll/027-050.html
Felgentreff K, Perez-Becker R, Speckmann C, Schwarz K, Kalwak K, Markelj G, Avcin T, Qasim W, Davies EG, Niehues T, Ehl S (2011) Clinical and imm nological manifestations of patients with atypical severe combined immunodeficiency. Clin Immunol 141:73-82
Grumach AS, Kirschfink M; (2014) Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. 2014 Oct;61(2):110-7
Jesenak M, Banovcin P, Jesenakova B, Babusikova E. Pulmonary manifestations of primary immunodeficiency disorders in children. Front Pediatr. 2014 Jul 25;2:77
Kersseboom, Brooks, Weemaes (2011) Educational Paper: Syndromatic forms of primary immunodeficiency. Eur J Pediatr. 170:295-308
Krudewig J, Baumann U, Bernuth von H, Borte M, Burk-hard-Meier U, Dueckers G, Foerster-Waldl E, Franke K, Habermehl P, Hönig M, Kern W, Kösters K, Kugel K, Lehrnbecher T, Liese J, Marks R, Müller GA, Müller R, Nadal D, Peter HH, Pfeiffer-Kascha D, Schneider M, Sitter H, Späth P, Wahn V, Welte T, Niehues T; Arbeitsgemeinschaft Pädiatrische Immunologie e. V. (API) (2012) [Interdisciplinary AWMF guideline for the treatment of primary antibody deficiencies]. Klin Padiatr 224:404-415
Martinon F, Aksentijevich I (2015) New players driving inflammation in monogenic autoinflammatory di- seases. Nat Rev Rheumatol. 2015 Jan;11(1):11-20
Platt C, Geha R, Chou J (2014) Gene hunting in the genomic era: Approaches to diagnostic dilemmas in patients with primary immunodeficiencies. J Allergy Clin Immunol 2014; 134:262-8
Relan M, Lehman HK. 2014 Common dermatologic manifestations of primary immune deficiencies. Curr Allergy Asthma Rep. 2014 Dec;14(12):
Rose CD, Neven B, Wouters C (2014) Granulomatous inflammation: The overlap of immune deficiency and inflammation. 28(2):191-212
Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, Schneider DT, Manfras B, Pannicke U, Willemze R, Knüchel R, Göbel U, Schulz A, Borkhardt A, Friedrich W, Schwarz K, Niehues T. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008 May 8;358(19):2030-8
Seger RA (2010) Chronic granulomatous disease: ecent advances in pathophysiology and treatment. Neth J Med 68:334-340
Uhlig HH, Schwerd T, Koletzko S, Shah N, Kammermeier J, Elkadri A, Ouahed J, Wilson DC, Travis SP, Turner D, Klein C, Snapper SB, Muise AM; COLORS in IBD Study Group and NEOPICS. Gastroenterol gy. 2014 Nov;147(5):990-1007
van der Burg M, Gennery AR (2011) Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur J Pediatr 170:561-571.