Nils Von Hentig, Frankfurt
Gewebegängigkeit der antiretroviralen Therapie
Die Gewebegängigkeit antiretroviraler Arzneimittel spielt eine große Rolle bei der HIV-Therapie. Nicht nur das Eindringen in periphere Zellen sichert den Behandlungserfolg, sondern auch die Penetration in Gewebe und Organe, in denen sich HIV über längere Zeit aufhält und repliziert.
Physiologie
Ist ein Stoff aus dem GI-Trakt aufgenommen, wird er mit der Blutzirkulation in alle Körperteile transportiert und tritt aus den Kapillaren ins periphere Gewebe über. Das Endothel peripherer Kapillaren hat, für den Austausch von Wasser und darin gelösten oder suspendierten Stoffen zwischen dem Blut und der extrazellulären Flüssigkeit des umliegenden Gewebes, Öffnungen von ca. 50 nm Durchmesser und Zwischenzellspalten von 0,1 bis 1 µm Weite. Um in einige Kompartimente zu gelangen, muss jedoch eine Blut-Gewebeschranke überwunden werden.
Blut-Gewebeschranken bestehen aus besonders „dichten“ Verbindungen (Tight Junctions) der Endothelzellen der Blutgefäße. Sie formieren einen („wasserdichten“) Abschluss des Gewebes gegenüber Noxen, die im Blutkreislauf zirkulieren.
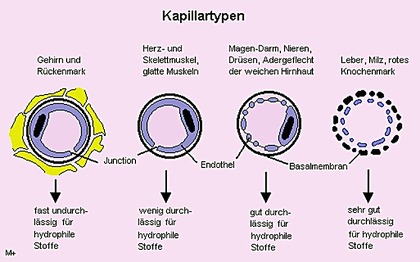
Abb. 1 Kapillartypen
Diese Unterschiede zeigen auch die Kapillartypen der Organe: Während Gehirn und Rückenmark fast komplett gegenüber den wasserlöslichen Stoffen im Blut abgeschlossen sind, lassen Organe, die für die Aufnahme und/oder Elimination von Arzneimitteln wichtig sind, chemische Verbindungen bestimmter Größe und Eigenschaften besonders leicht passieren (Abb. 1).
Endothelzellen sind mit 0,3-0,5 µm äußerst dünn. Enterozyten, d.h. die Epithelzellen des Darmes, sind im Vergleich dazu mit 17-30 µm erheblich dicker. Das Verhältnis von Cholesterin zu Phospholipiden liegt, wie bei anderen Endothelzellen auch, bei 0,7.
Blut-Hirn-Schranke
Reine
Diffusionsprozesse durch die Zellmembran sind daher über die
Blut-Hirn-Schranke schnell. Die Endothelzellen weisen auf ihrer
Zellmembran für die Regulation des Wasserhaushaltes des Gehirnes
eine Vielzahl von sogenannten Aquaporinen auf. Diese Kanäle
ermög-lichen Wassermolekülen die freie Diffusion sowohl in Richtung
Gehirn als auch zum Blut.
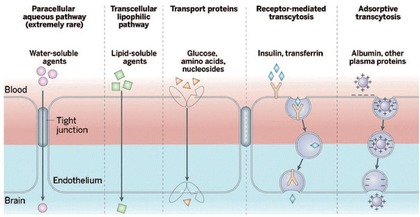
Abb. 2 Die endotheliale Blut-Hirn-Schranke
Die
einfachste Form des Transportes durch die Blut-Hirn-Schranke stellt
der passive Transport (Diffusion) dar, welcher sowohl durch die
Zellmembran der Endothelien als auch durch die Tight Junctions
stattfinden kann. Dabei wird – wie bei jeder Diffusion – ein
Konzentrationsausgleich oder der Ausgleich eines elektrochemischen
Gradienten angestrebt. Die lipophilen Eigenschaften der Zellmembran
und ihre dichte Verknüpfung über die Tight Junctions reduzieren
jedoch die Zahl der Substanzen, die durch Diffusion die
Blut-Hirn-Schranke überwinden können. Die Durchlässigkeit der
Blut-Gewebe-Schranken für ein bestimmtes Molekül steht in direktem
Verhältnis zu seiner
Lipophilie. Bezüglich der Masse verhält
es sich umgekehrt proportional. Das heißt, je lipophiler und kleiner
eine Verbindung ist, umso leichter kann sie durch das Endothel
hindurch diffundieren (Abb. 2).
Allerdings könne Substanzen auch Blut-Gewebeschranken überwinden, wenn sie Substrat dort vorhandener Transporter sind (Abb. 3).
Weiterhin sind Arzneistoffe im Blut in unterschiedlichem Ausmaß an Plasma-Eiweiße gebunden. Nur der freie Anteil kann jedoch in die Zielgewebe der Therapie eindringen.
Pharmakologie
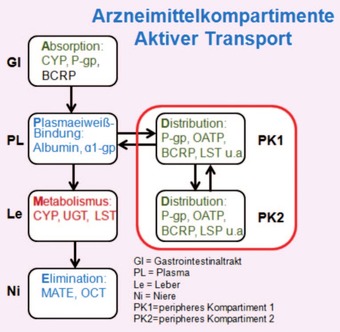
Abb. 3 Aktiver Transport (Transportersysteme) in verschiedene Arzneimittelkompartimente
Arzneimittel können verschiedene Wege gehen, um diese Barrieren zu überwinden. Je kleiner das Molekülgewicht, je höher die Polarität, je geringer die Plasma-Eiweißbindung, desto höher sind die im Zielgewebe zu erreichenden Konzentrationen. Hinzu kommen der Umgang mit aktiven Transportsystemen, d.h. eine hohe Affinität zu zellulären Influxtransportern und eine niedrige Affinität zu Effluxtransportern bzw. deren aktive Hemmung erhöhen die Gewebekonzentration der Arzneistoffe. Transportersysteme unterliegen hierbei sowohl einer möglichen Sättigungskinetik oder einer genetischen Varianz, d.h. sind in der Lage, immer nur eine bestimmte Menge AM/Zeit zu transportieren.
Umgekehrt
schützen sich das ZNS oder die Fortpflanzungsorgane, wie der Hoden,
vor Noxen, die aus Blut auf-
genommen werden, auch Tumorzellen
oder Stammzellen schützen sich so vor zelltoxischen Substanzen:
Viele Arzneimittel werden als solche erkannt und folglich entweder
nicht ins Gewebe aufgenommen oder nach der für die Zelle
„ungeplanten“ Aufnahme aus dieser gleich wieder ausgeschleust.
Dabei spielen zelluläre Transporter wie ABCB1 (P-Glykoprotein), BCRP
(Breast-Cancer Resistance Protein), OATP u.a. eine wichtige Rolle
(Abb. 3).
Charakteristika der ARVs
Die Charakteristiken der ARVs spielen bei der (aktiven) Überwindung von Blut-Gewebe-Schranken hierbei eine große Rolle (Abb. 4). Die Charakteristiken der ARVs spielen hierbei eine große Rolle (Abb. 4). Grundsätzlich gilt, wie bereits kurz erwähnt:
Je geringer (1) die Plasmaeiweißbindung, (2) das Molekülgewicht, (3) die Affinität zu Effluxtransportern und je höher (4) die Ionisation/Ladung, (5) die Affinität zu Influxtransportern, (6) der Anteil intrazellulärer Aktivierung, desto höher sind die Gewebekonzentrationen. Und je größer das (7) therapeutische Fenster, d.h. das Überschreiten der Hemmkonzentration des Virus desto besser ist die Wirksamkeit.
| Proteinbindung: | NNRTI ≥PI ≥INI >CCR5 >NRTI |
| Molekülgewicht: | PI >CCR5 >INI >NNRTI >NRTI |
| Lipophilität: | NNRTIs >PI >NRTI |
| Ionisation/Ladung: | Tenofovir |
| Transmembranäre Transporter: | PI, INI: P-Glykoproteinsubstrate NRTI: OATP-Substrate |
| Großes therapeutisches Fenster: | DGV >LPV/r >DRV/r >EFV |
| Intrazelluläre Aktivierung/HWZ: | TAF >TDF >andere NRTI |
| NNRTI= Nicht-nukleosidale reverse Transkriptase
Inhibitoren; PI= Proteaseinhibitoren; INI= Intergase-Inhibitoren; CCR5= CCR5-Inhibitoren; NRTI= Nukleos(t)idale Reverse Transkriptase-Inhibitoren; DGV= Dolutegravir; LPV/r= Lopinavir/r; DRV= Darunavir/r; EFV= Efavirenz; TAF= Tenofoviralefenamid; TVD= Emtricitabine/Tenofovirdisoproxilfumarat. |
|
Tab. 1 Die Substanzeigenschaften beeinflussen Verteilung/Wirksamkeit im ZNS
Ob Lipophilität (Fettlöslichkeit) oder Hydrophilität (Wasserlöslichkeit) gefragt sind, hängt dabei vom Ziel des Arzneimittels ab, das erreicht werden soll bzw. der Barriere, die überwunden werden muss (Tab. 1).
Octanol-Wasser-Verteilungskoeffizient
In der Pharmazie wird die Lipophilität/Hydrophilität mithilfe des Octanol-Wasser-Verteilungskoeffizienten bezeichnet. Grundsätzlich gilt: Je kleiner der Koeffizient, desto hydrophiler (z.B. Tenofovir), je größer desto lipophiler (z.B. Efavirenz). Grundsätzlich ist es so, dass lipophile Substanzen besser die Blut-Hirn-Schranke überwinden können, während sich z.B. Tenofovir-DF besser für eine PrEP im wässrigen Anal- oder Vaginalmilieu eignet.
Plasma-Eiweiß-Bindung
| ARV | Molekülgewicht | Proteinbindung (%) |
|---|---|---|
| NRTI
| ||
| Zidovudin | 267.2 | 34-38 |
| Lamivudin | 229.3 | >36 |
| Abacavir | 286.3 | 49.0 |
| Emtricitabin | 247.2 | <4.0 |
| Tenofovir | 305.2 | <0.7 |
| NNRTI
| ||
| Nevirapin | 266.3 | 60.0 |
| Efavirenz | 315.7 | 99.5 |
| Etravirine | 435.0 | 99.9 |
| Rilpivirine | 402.9 | 99.7 |
| PI
| ||
| Ritonavir | 721.0 | 98-99 |
| Cobicistat | 776.0 | 97-98 |
| Lopinavir | 628,8 | 98-99 |
| Saquinavir | 670.9 | 98.0 |
| Atazanavir | 704.9 | ++(+) |
| Darunavir | 548.0 | 95.0 |
| CCR5-Inhibitor
| ||
| Maraviroc | 514.0 | 76.0 |
| INI
| ||
| Raltegravir | 444,0 | 83.0 |
| Dolutegravir | 441.4 | ≥98.9 |
| Elvitegravir | 447.9 | 98-99 |
Tab. 2 Molekülgewicht und Ausmaß der
Plasmaeiweißbindung antiretroviraler Substanzen
Arzneistoffe unterliegen in unterschiedlichem Ausmaß der Plasmaeiweißbindung d.h. einer reversiblen Bindung an Eiweißbestandteile des Blutes (hauptsächlich Albumin, aber auch a1-Globuline). An dieser Proteinbindung können verschiedene Formen chemischer Bindungen beteiligt sein, wie z.B. Ionenbindungen, H-Brückenbindungen oder Wechselwirkungen. Die Plasmaeiweißbindung bedeutet, dass nur mehr der freie, ungebundene Anteil des Arzneistoffes mit der Umgebung reagieren kann, d.h. eine Arzneimittelwirkung entfaltet. Der durch Proteine gebundene Anteil eines Stoffes ist pharmakologisch inaktiv. Die Plasmaeiweißbindung kann auch die Halbwertszeit mancher Arzneistoffe verlängern, da deren Ausscheidung über die Niere nur verlangsamt erfolgen kann. Zu bekannten Substanzen, die zu einem großen Teil im Plasma an Proteine gebunden sind, gehören z.B. Sulfonamide, Sulfonylharnstoffe, ASS, Phenprocoumon, Warfarin, aber auch verschiedene antiretrovirale Arzneimittel (Tab.2).
Auch die Molekülgröße spielt eine Rolle bei der Aufnahme in das Gewebe bzw. der Passage verschiedener Blut-Organschranken, z.B. ins ZNS, der Plazenta oder in die Milchgänge stillender Mütter: Moleküle <400 D penetrieren fast jedes Gewebe, größere sind auf Transporter angewiesen; Moleküle >1.000 D werden nicht mehr oral aufgenommen und sind auf aktiven Transport angewiesen.
Transmembranärer Transport
Über den Transport von Arzneimitteln über zelluläre Barrieren hinweg ist bereits im ersten Teil dieser Reihe geschrieben worden. Dennoch macht es Sinn, im Zusammenhang mit der Gewebegängigkeit einige dieser Systeme, die Influx-Transporter, nochmals näher zu betrachten.
Zelluläre Influx-Transporter
| Patienten-ID | Substanz | Plasma (ng/mL) | Hoden (ng/mL) |
|---|---|---|---|
| 6 | DRV
RTV | 2.043.7
48.9 | 396.6
389.3 |
| 7 | TDF
FTC | 61.7
214.4 | 44.7
251.8 |
| 8 | 3TC
EFV | 207.8
7.448.9 | 147.1
1.856.4 |
| 9 | 3TC DRV RTV | 125.8 2.375.8 236.0 | 140.4
523.1 683.8 |
| 10 | TDF
3TC ATV RTV | 51.0
136.7 1.300.2 271.8 | 43.5 21.3 1.029.8 523.8 |
http://www.croiconference.org/sites/default/
files/posters-2015/534.pdf
CROI, Huang et al. 2014
Tab. 3 ARV Konzentrationen in Patienten in
Plasma und Hoden. Die Hoden-Konzentration
wird als ng/mL ausgedrückt, wobei angenommen wird, dass 1mL ~ 1gr Gewebe entspricht.
OATP und PGT
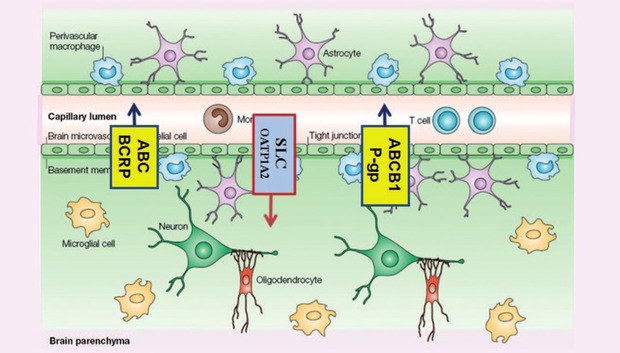
OATP kommt in vielen Körpergeweben vor, so z.B. in der Leber: SLCO1B1, SLCO1B3, den Nieren: OATP-H (SLCO4C1), dem ZNS: OATP-A (SLCO1A2), OATP-F, den Hoden: OATP-I (SLCO6A1), SLCO2B1 sowie überall: PGT (SLCO2A1), OATP-B (SLCO2B1), OATP-D (SLCO3A1), OATP-E (SLCO4A1).
Blut-Hoden-Schranke
Hier wird klar, dass einige ARVs, wie z.B. RTV, eine hohe Affinität zu Influx-Transportern haben. Andere, wie DRV, ATV oder EFV zeigen diese Eigenschaft nicht. NRTI, wie 3TC, TDF und FTC zeigen eine Plasma/Hoden-Ratio von annähernd 1 (Tab. 3).
Blut-Hirn-Schranke
Ein weiteres Beispiel ist die Blut-Hirn-Schranke, auch hier dient OATP1A2 als Influx-Transporter (Abb. 4).
Dass beim Zell-Influx verschiedene Arzneistoffe um dieselben Transporter konkurrieren, macht die Situation kompliziert und eine Vorhersage der klinischen Interaktion schwierig. Wichtig ist aber, dass grundsätzlich eine Abschwächung der Gewebegängigkeit zweier AM, die über denselben Weg transportiert werden, aufgrund einer Sättigungskinetik des Transporters auftreten kann (Tab. 3).