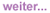Nils Von Hentig, Frankfurt
Pharmakologie der HIV-Therapie
Prinzipien der Arzneimittelinteraktionen
Arzneimittelinteraktionen beschränken sich nicht auf die bekannten Stoffwechselwege in Leber und Niere. Auch Wechselwirkungen am Darmepithel bei der Aufnahme von Substanzen, bei Transportwegen über die Membranen der Zielzellen bis hin zur gegenseitigen Verdrängung aus der Plasmaeiweißbindung spielen eine Rolle. Somit sind innerhalb der antiretroviralen Therapie als auch in Kombination mit weiteren Medikamenten Interaktionen auf mehreren Ebenen gleichzeitig möglich.
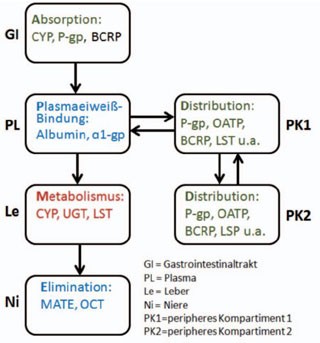
Abb. 1
Absorption und First-Pass
Grundsätzlich können Arzneimittelinteraktionen bereits bei der Absorption von Arzneimitteln im Magen-Darm-Trakt ablaufen. Diese treten vor allem bei sogenannten fast-extracting drugs, d.h. Medikamenten, die bereits beginnend über die Magenschleimhaut aufgenommen werden, wie z.B. Nelfinavir, Saquinavir und Atazanavir. Hier entscheidet zum einen der pH-Wert des Magens über die Resorption, wie z.B. im Fall von Atazanavir bei gleichzeitiger chronischer Einnahme von Protonenpumpenhemmern14-17, zum anderen können parallel eingenommen Metallkomplexbildner, wie z.B. Maaloxan, die Absorption von Wirkstoffen durch Bildung unlöslicher Komplexe unmöglich machen.18, 19
Desweiteren folgen Interaktionen bei der Aufnahme über die Darmwand, dort sind bereits Cytochromoxidasen lokalisiert, d.h. Enzyme des first-pass-Stoffwechsels, die sich im weiteren Stoffwechselweg vor allem in der Leber finden sowie verschiedene zelluläre Effluxtransporter, beispielsweise P-Glykoprotein (P-gp) und Breast Cancer Resistance Protein (BCRP).
Arzneimittelinteraktionen treten bereits bei der Absorption von Arzneimitteln im Magen-Darm-Trakt auf, wobei zum einen der pH-Wert (verändert durch PPI), zum anderen Interaktionen mit Komplexbildnern eine Rolle spielen (z.B. zweiwertige Al oder Mg-Kationen). Es folgen mögliche Interaktionen bei der Aufnahme über die Darmwand, dort sind bereits Cytochromoxidasen lokalisiert sowie verschiedene zelluläre Effluxtransporter (z.B. P-gp, BCRP).
Arzneimittelinteraktionen in der Leber
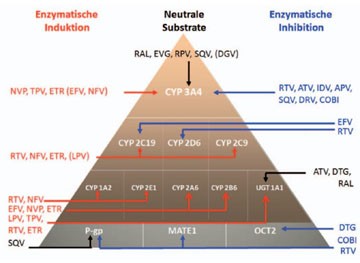
Abb.
2 Hinzu kommt, dass das Ausmaß des Effektes auf die betroffenen CYP
von ARV zu ARV variiert. Letztlich ist die Konzentration am Enzym
entscheidend, ob und in welchem Ausmaß ein Effekt (Hemmung oder
Induktion) erreicht wird (siehe Abb. 3).

Abb. 3
Arzneimittelinteraktionen
zwischen einzelnen antiretroviralen Substanzen bzw. mit der
Begleitmedikation können in besonders niedrigen oder toxisch hohen
Arzneimittelspiegeln resultieren. Alle PI, NNRTI sowie der
CCR5-Hemmer,
Maraviroc
werden bei der ersten Passage durch die Leber über Cytochrom P450
(CYP), insbesondere CYP3A, verstoffwechselt. Ritonavir, Cobicistat
und Atazanavir sind dabei potente Inhibitoren, während
beispielsweise Nevirapin und Tipranavir Induktoren desselben Systems
sind. Internetseiten geben entsprechende Informationen und
Handlungsanweisungen für den Fall vermuteter Interaktionen innerhalb
der cART (z.B. www.hiv-druginteractions.org/).
Arzneimittelinteraktionen
von PI und NNRTI, beispielsweise mit Antazida, Lipidsenkern,
Psychopharmaka, Antiepileptika, Opiaten, Anti-HCV-Medikamenten,
Chemotherapeutika, Immunsuppressiva und neuen Antikoagulanzien
spielen in der Klinik eine bedeutende Rolle. Bis zu 50% der
HIV-Patienten nehmen zumindest zeitweise H2-Rezeptorenblocker oder
Protonenpumpeninhibitoren ein, welche z.B. die Resorption von
Indinavir,
Nelfinavir, Atazanavir oder Rilpivirine20
hemmen. Gleiches gilt für Maraviroc: Dosierungsanweisungen
entsprechend der eingesetzten Komedikation deuten auf das
Interaktionspotential hin.
Tab. 1
Alle PI, NNRTI sowie Maraviroc werden bei der Leberpassage über Cytochrom P450 (CYP), insbesondere CYP3A, verstoffwechselt. Ritonavir, Cobicistat und Atazanavir sind dabei potente Inhibitoren, während beispielsweise Nevirapin und Tipranavir Induktoren desselben Systems sind.
Cytochromoxidasen
Cytochromoxidasen sind Enzyme, die in verschiedenen Zellen des menschlichen Körpers vorkommen. Eine wichtige Rolle spielen hierbei die CYPs, die im Darmepithel und in Hepatozyten lokalisiert sind, also Zellschichten, über welche Arzneimittel hinweg in den Körper aufgenommen bzw. verstoffwechselt und abgebaut werden. ARVs können die Aktivität bzw. Expression der CYPs entweder hemmen oder induzieren, einige ARVs sind ausschließlich Substrate dieser Oxidasen. Da sich in einer cART immer mehrere auf die CYPs einwirkende Arzneistoffe befinden, kommt es letztlich auf den sogenannten Netto-Effekt an, der angibt, welche der eingesetzten Substanzen in welchem Ausmaß von Induktion oder Inhibition betroffen sind.
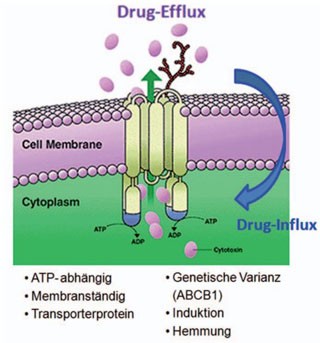
Abb. 4 Eine ARVs sind Substrate, andere wiederum Inhibitoren oder Induktoren von P-Glykoprotein
Das folgende Schaubild zeigt den Einfluss verschiedener ARVs auf die Cytochromoxidasen im sogenannten steady-state. Ca. 2-4 Wochen nach Therapiebeginn wird ein Fließgleichgewicht in diesem System erreicht, wenn die Induktion der CYP-Expression abgeschlossen und die Induktions- oder Hemmwirkung zu 100% erreicht wird. Zusätzlich weisen einige CYPs genetische Polymorphismen auf, die das Ausmaß ihrer Expression determinieren.
ARVs können ausschließlich Substrate, andere Induktoren oder Inhibitoren der Cytochromoxidasen sein. Dabei variiert das Ausmaß des Effektes auf die betroffenen CYP von ARV zu ARV, letztlich abhängig von der jeweiligen Konzentration am Enzym. In einer cART kommt es auf den sogenannten Netto-Effekt an, der angibt, welche der eingesetzten Substanzen in welchem Ausmaß von Induktion oder Inhibition betroffen sind.
Zelluläre Transportwege
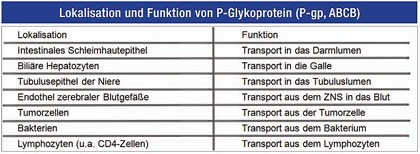
Viele ARVs sind Substrate oder Inhibitoren von P-Glykoprotein, einem zellulären Effluxtransporter, der sich an einigen Transportwegen aus dem Blut in Zielgewebe wieder findet.
P-Glykoprotein ist ein Membranständiges Transportprotein, welches ATP-abhängig Toxine oder auch Arzneistoffe, die von der Zelle als Toxine angesehen werden, nach dem Eindringen ins Zielgewebe aktiv aus diesem wieder ausschleust, d.h. einer therapeutischen Wirkung entgegen arbeitet. Die P-Glykoprotein-Expression wird über das MDR-1 Gen codiert, welches eine bedeutende genetische Varianz in Patienten aufweist. Die neue Nomenklatur der Proteine weist P-Glykoprotein als Mitglied der ABC-Transporter-Familie aus (ABCB1).
Durch Hemmung von P-Glykoprotein mittels Ritonavir an der Bluthirnschranke kann es zur Etablierung von antiretroviral wirksamen ZNS-Spiegeln von ansonsten nur schlecht liquorgängigen Proteaseinhibitoren führen. Derzeit läuft eine Studie der Arbeitsgruppe von Scott Letendre in den U.S.A. um die Frage zu klären, ob derselbe Effekt durch Cobicistat erreicht werden kann (www.fda.gov, clinical trials Identifier No. NCT02251236). Ergebnisse dieser Studie liegen noch nicht vor.
P-Glykoprotein ist ein Membranständiges Transportprotein, welches ATP-abhängig Arzneistoffe nach dem Eindringen ins Zielgewebe aktiv aus diesem wieder ausschleust. Die P-Glykoprotein-Expression wird über das MDR-1 Gen codiert. P-gp findet sich an den verschiedensten Gewebeschranken des Organismus, u.a. in Darm, Leber, Niere und ZNS.
Andere zelluläre Transporter
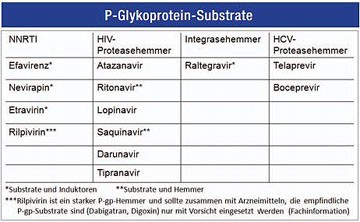
Tab. 2
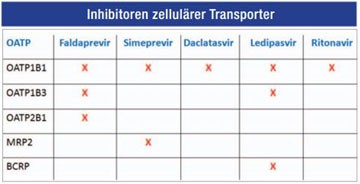
Tab. 3
Zunehmend an Bedeutung gewinnen andere zelluläre Transporter, die in verschiedenen Geweben des Körpers lokalisiert sind. Insbesondere neue HCV-Medikamente sind außerhalb ihres Stoffwechsels über CYP, UGT oder P-gp auch noch Substrate von OATP oder BCRP. BCRP ist u.a. im Dünndarmepthel, der Blut-Hoden oder Blut-Hirnschranke, der Plazenta oder den Milchdrüsen lokalisiert. BCRP kann entweder den Übertritt von Arzneimitteln in bestimmte Kompartimente (z.B. ZNS, Fetus) verhindern oder Stoffe abgeben, wie beispielsweise Biotin und Roboflavin in die Muttermilch.
OATP hingegen findet sich in der Leber: LST-1 (SLCO1B1), LST-2 (SLCO1B3), den Nieren: OATP-H (SLCO4C1), dem ZNS: OATP-A (SLCO1A2), OATP-F (und in Leydig-Zellen, SLCO1C1), den Hoden: OATP-I (SLCO6A1) bzw. ubiquitär: PGT (SLCO2A1), OATP-B (SLCO2B1), OATP-D (SLCO 3A1), OATP-E (SLCO4A1). Neuere HCV-Medikamente, wie Sofosbuvir oder Ledipasvir aber auch Ritonavir können OATP hemmen.
Klinische Relevanz haben auch andere zelluläre Transporter, die in verschiedenen Geweben des Körpers lokalisiert sind. Viele Medikamente, z.B. neue HCV-Therapeutika, sind neben CYP, UGT oder P-gp auch noch Substrate von OATP oder BCRP.
Renale Transporter
Auch renale Transporter spielen eine Rolle im Stoffwechsel von Arzneimitteln bzw. körpereigenen Stoffen, wie z.B. Kreatinin. Das Schaubild verdeutlicht den Einfluss bestimmter ARVs auf MATE oder OCT, zwei transmembranäre Transporter, welche an der Ausscheidung von Kreatinin über die Tubuluszellen in den Urin beteiligt sind.
Eine Hemmung dieser Transporter führt zu einem messbar erhöhten Serum-Kreatininwert mit in der Folge falsch niedriger errechneter Glomerulärer Filtrationsrate (GFR nach Cockroft-Gaullt), nicht aber zu einer realen Einschränkung der Nierenleistung.
Auch wenn beispielsweise die gleichzeitige Gabe von Dolutegravir, einem OCT2-Hemmer und Metformin, einem OCT2-Substrat zu erhöhten Metforminspiegeln führt, so zeigte keiner der 41 Patienten in einer Zulassungsstudie mit Dolutegravir, welche auch Metformin einnahmen, vermehrt Nebenwirkungen oder musste die Dosis von Metformin anpassen. Die Fachinformation weist jedoch auf diese Möglichkeit hin.
Die Bedeutung mögliche klinische Relevanz renaler zellulärer Transporter bei der tubulären Sekretion von Arzneimitteln wird zunehmend verstanden, beispielsweise bei der Hemmung von OCT2 (Metformin-Transport) durch Dolutegravir.
Plasmaeiweißbindung
Jüngste Daten hinsichtlich der unerwarteten Interaktionen zwischen HIV- und Erstgenerations-HCV-Proteaseinhibitoren weisen darauf hin, dass auch die gegenseitige Verdrängung aus der Plasmaeiweißbindung eine Rolle spielen kann. Wenn zwei, normalerweise in hohem Masse an Plasmaeiweiß gebundene Substanzen, um eben diese Bindung konkurrieren, resultiert das in einer größeren Menge ungebundener Arzneistoffe, welche dann frei werden für Abbau und Ausscheidung. Somit sinken die Plasmakonzentrationen beider bzw. derjenigen Substanz deren Bindung zu Plasmaeiweiß schwächer ist.21
Wenn zwei in hohem Maße an Plasmaeiweiß gebundene Substanzen um diese Bindung konkurrieren, resultiert das in einer beschleunigten Elimination beider Substanzen. Die wirksamen Plasmakonzentrationen sinken.
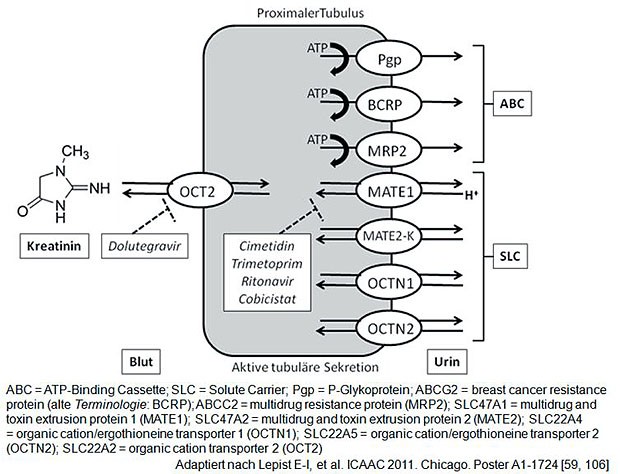
Abb. 5
Therapeutisches Drug Monitoring
Therapeutisches Drug Monitoring (TDM) ist ein möglicher Weg, Arzneimittelkonzentrationen zu messen.22
Die Plasmaspiegel-Messung antiretroviraler Arzneimittel im Rahmen des Therapeutischen Drug Monitoring schließt verschiedene Parameter ein:
Mithilfe der Minimalkonzentration im Dosis-Intervall, Cmin, bzw. der Wirkspiegel vor erneuter Arzneimittelgabe, Ctrough, wird ein Unterschreiten therapeutischer Wirkspiegel überprüft. Die Maximalkonzentration im Dosis-Intervall, Cmax, gibt Hinweise auf unerwünschte Wirkungen, die durch sehr hohe Arzneimittelspiegel bedingt sein können. Die Gesamtexposition eines Arzneimittels, die Fläche unter Konzentrations-Zeitkurve, AUC (Area Under the time-concentration Curve), kann sowohl mit der Wirksamkeit als auch Nebenwirkungen korreliert sein.
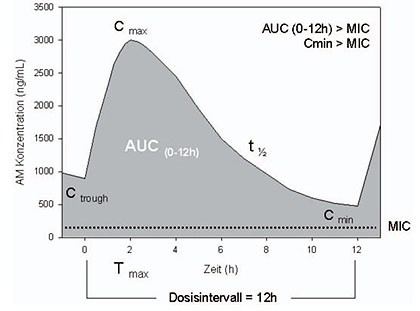
Abb. 6
Generell werden bisher nur HIV-Protease Inhibitoren (PI), Nicht Nukleosidale Reverse Transkriptase Inhibitoren (NNRTI) und der CCR5-Inhibitor Maraviroc routinemäßig in Plasma bzw. Serum gemessen. Neuere Substanzen, wie Elvitegravir bzw. Dolutegravir werden ebenfalls in die Routine aufgenommen werden. Allen Gemeinsam ist, dass deren Wirksamkeit Cmin-abhängig ist, das heißt eine minimale wirksame Konzentration im Dosis-Intervall nicht unterschritten werden soll.
Nukleosidale Reverse Transkriptase Inhibitoren (NRTI) hingegen sind prodrugs und werden nach Resorption aus dem Magen-Darm-Trakt erst in der Zielzelle, mehrfach phosphoryliert, antiretroviral wirksam. Ein Zusammenhang zwischen Plasmakonzentration der prodrugs und intrazellulären Konzentrationen der wirksamen Substanzen besteht nicht, und sie werden deshalb auch nicht im Rahmen des Routine-TDM gemessen.
Für PI und NNRTI hingegen gibt es Empfehlungen für minimale bzw. im Patienten angestrebte effektive Konzentrationen im Plasma, welche entweder in Studien mit Therapie-naiven Patienten oder aus in-vitro-Daten generiert und extrapoliert wurden.18
Beim TDM von PI, NNRTI und Maraviroc werden meist Cmin mit der Wirkung und Cmax mit Nebenwirkungen korreliert. Für Integrasehemmer ist noch keine sichere Konzentrations-Wirkungsabhängigkeit etabliert, es gibt allerdings neben PI und NNRTI auch für Raltegravir empfohlene effektive Plasmakonzentrationen für die Behandlung von Wildtyp HIV-1.
Konsequenzen für Klinik und Praxis
Der Talspiegel, Ctrough, wird in der Regel für die Überprüfung der Patienten-Compliance bzw. vermutete Resorptionsstörungen benutzt. Wiederholte Ctrough-Messungen dienen der Bestimmung der intra-individuellen Variabilität der Arzneimittelspiegel. Auch die Ratio aus Ctrough und Anzahl der vorhandenen genotypischen Resistenzmutationen (gIQ) bzw. deren Phänotyp (phenotypic Inhibitory Quotient, pIQ) gibt Hinweise auf potentiell zu niedrige Arzneimittelspiegel bei vorbehandelten Patienten. Dabei ist zu beachten, dass Ctrough Cmin ist. Die Minimalkonzentration im Dosis-Intervall tritt bei vielen Patienten möglicherweise erst 1, 2 oder 4 Stunden nach Einnahme der Medikation auf, insbesondere bei verzögerter Resorption, z.B. in der Schwangerschaft.
Die Maximalkonzentration, Cmax, wird benutzt, um einen Hinweis auf die Ursache für mögliche Toxizitäten zu bekommen. Der Zeitpunkt der Cmax, Tmax, liegt i.d.R. zwischen 1 und 6 Stunden, je nach cART, so dass eine Messung um diesen Zeitpunkt herum geplant werden sollte, wenn sich die Frage nach hohen Arzneimittelspiegeln stellt. Bisher existieren keine Normkurven für verschiedene Populationen, an Hand derer sich Cmax aus den zu anderen Zeitpunkten gemessenen Konzentrationen berechnen ließen.
Soll die Halbwertzeit, t1/2, ermittelt werden, um beispielsweise eine Kumulation von einzelnen Substanzen frühzeitig aufzudecken, so muss das gesamte Dosis-Intervall gemessen und vor allem die späte Phase erfasst werden.
Zur Bestimmung der vollständigen Pharmakokinetik über das Dosis-Intervall im erwachsenen Patienten (einschließlich Konzentrations-Zeitkurve, AUC, oder tota-ler Clearance, Cltotal) haben sich die Messzeitpunkte 0, 1, 2, 4, 6, 9, 12 (24) Stunden bewährt. Bei kleineren Kindern reichen mindestens die Messzeitpunkte 0, 2, 4, 8, 12 Sunden aus. Die Konzentrationsbestimmung im sensitiven Tandem-Massenspektrometer (LC-MS/MS) kommt hierbei mit jeweils weniger als 0.5 mL Plasma aus.
Generell ist es hilfreich, das Vorgehen in der Praxis zu standardisieren, um die Daten vergleichend interpretieren zu können. Die Messung sollte frühestens zwei Wochen nach Beginn der Therapie (im steady state, d.h. wenn die verstoffwechselnden Enzymsysteme konstant funktionieren) stattfinden. Der Zeitraum seit der letzten Einnahme der cART bis zur Messung muss dokumentiert werden sowie generell alle Begleitmedikamente. Patienten sollen am PK-Tag gewogen, ein Resistenztest sowie die CD4-Zellzahlbestimmung wenn möglich zeitnah eingeplant werden und die Probenanalyse sollte in einem extern Qualitätskontrollierten Labor stattfinden (z.B. durch INSTAND e.V., Düsseldorf oder KKGT, Nijmwegen, Niederlande).23, 24
Es ist für die heutige HIV-Behandlung unablässig ein Grundverständnis für pharmakologische Prinzipien bei Arzneimittelinteraktionen zu entwickeln. Zusätzlich sollte, wo TDM möglich ist, dieses richtig eingesetzt werden.
Bei direkten Fragen zu vermuteten Arzneimittelinteraktionen gibt es entsprechende Internet-basierte Beratungs-Angebote in deutscher Sprache (z.B. www.ifi-interaktions-hotline.de, http://www.inxfo.de/
Der Talspiegel, Ctrough, dient der Überprüfung der Patienten-Compliance bzw. vermuteter Resorptionsstörungen. Die Ratio aus Ctrough und der Anzahl vorhandener genotypischer Resistenzmutationen (gIQ) kann Hinweise auf potentiell zu niedrige Arzneimittelspiegel bei vorbehandelten Patienten geben. Ctrough Cmin, die Minimalkonzentration im Dosis-Intervall tritt bei vielen Patienten erst 1, 2 oder 4 Stunden nach Einnahme der Medikation auf. Diese Verzögerung ist insbesondere in der Schwangerschaft beschrieben. Die Maximalkonzentration, Cmax, lässt sich mit möglichen Nebenwirkungen korrelieren. Der Zeitpunkt der Cmax, Tmax, liegt i.d.R. zwischen 1 und 6 Stunden. TDM sollte frühestens zwei Wochen nach Beginn der Therapie (im steady state) stattfinden.
Pharmakogenetik
Die genetische Disposition von Patienten in Bezug auf den Arzneimittelstoffwechsel spielt bisher im klinischen Alltag der antiretroviralen Therapie eine untergeordnete Rolle. Im Jahr 2002 konnten Fellay et al. zeigen, dass Polymorphismen des MDR-1 Gens einen direkten Einfluss auf den Erfolg einer Therapie mit dem Proteaseinhibitor Nelfinavir haben, welcher Substrat von P-Glykoprotein ist.25 Das MDR-1 Gen codiert für die P-Glykoprotein-Aktivität, einer Effluxpumpe, welche Arzneimittel aus vielen Zellen ausschleust. Patienten mit dem MDR-1 3435 TT-Subtyp hatten signifikant höhere Plasmaspiegel und einen stärkeren Anstieg der CD4 Zellen unter Therapie als Vergleichspatienten mit den MDR-1 3435 CC- oder CT-Subtypen. Die Arbeit von Fellay et al. konnte jedoch bis heute nicht in anderen HIV-Therapieregimen reproduziert werden.
Ein Raltegravir betreffender Polymorphismus kodiert für die Aktivität von Urunosyl-Glukuronyl-Transferase A1 (UGT1A1), welche Bilirubin zu einer wasserlöslichen Substanz konjugiert, aber auch Raltegravir oder Morphin zu den jeweiligen Glukuroniden metabolisiert. Interaktionen mit Hemmern der UGT, wie z.B. Atazanavir spielen dennoch klinisch bisher keine Rolle, da erhöhte Raltegravir-Konzentrationen nicht mit dem Auftreten bestimmter Nebenwirkungen korreliert werden konnten. Jüngst publizierte Daten deuteten auf keinen relevanten Einfluss veränderter Rezeptorgenetik auf Raltegravir Plasmakonzentrationen. Die herabgesetzte Formation von Glukuronid-Metaboliten26 sowie die Steigerung der Plasmakonzentrationen um 41-92% bei Vorliegen eines UGT1A1*28/*28 Genotyps im Vergleich zu UGT1A1*1/*1 hatten keinerlei klinische Relevanz.27, 28
Die Erhöhung des indirekten Bilirubins durch die Atazanavir-induzierte, kompetitive UDP-Glucuronosyltransferase (UGT)-Inhibition ist sowohl Konzentrationsabhängig als durch die genetische Ausprägung dieses Enzyms bestimmt.
Zwei Arbeiten aus dem Deutschen Kompetenznetzwerk HIV/AIDS konnten zeigen, dass die Integration von Pharmakogenetik und Populationspharmakokinetik eine individualisierte Dosisfindung für Nevirapin ermöglicht29 bzw. dass Cytochrom P450 2B6- und CAR-Polymorphismen mit dem früheren virologischen Versagen einer Efavirenz-basierten cART korrelieren.30 Auf Empfehlungen hinsichtlich der pharmakogenetischen Testung im klinischen Alltag wird, auch in den Leitlinien der DAIG zur cART in Erwachsenen, zunächst jedoch verzichtet.31
Die genetische Disposition von Patienten in Bezug auf den Arzneimittelstoffwechsel spielt im klinischen Alltag der cART bisher eine untergeordnete Rolle. Auf Empfehlungen hinsichtlich der pharmakogenetischen Testung (Ausnahme HLA-B5701) im klinischen Alltag wird, auch in den Leitlinien der DAIG zur cART in Erwachsenen, zunächst verzichtet.31
Zusammenfassung
Die Pharmakologie der HIV Therapie ist komplex. Sind mehr als drei miteinander interagierende Substanzen Teil der Arzneimitteltherapie, so ist eine Vorhersage der Effekte sehr schwierig. Das standardisierte Therapeutische Drug Monitoring hilft, unerwartete Effekte frühzeitig zu erkennen. Eine Besonderheit stellt die zunehmend erforschte Zahl zellulärer Transporter dar, welche mit der HIV oder HCV-Therapie interferieren können.
1 Lee L, Karon J, Selik R, Neal J, Fleming P. Survival after AIDS diagnosis in adolescents and adults during the treatment era, United States, 1984-1997. JAMA 1998,285:1308-1315.
2 Mocroft A, Ledergerber B, Katlama C, Kirk O, Reiss P, d‘Arminio Monforte A, et al. Decline in the AIDS and death rates in the EuroSIDA study: an observational study. Lancet 2003,362:22-29.
3 Collot-Teixeira S, De Lorenzo F, Waters L, Fletcher C, Back D, Mandalia S, et al. Impact of different low-dose ritonavir regimens on lipids, CD36, and adipophilin expression. Clin Pharmacol Ther 2009,85:375-378.
4 Carr A. Cardiovascular risk factors in HIV-infected patients. J Acquir Immune Defic Syndr 2003,34 Suppl 1:S73-78.
5 Sax PE, Kumar P. Tolerability and safety of HIV protease inhibitors in adults. J Acquir Immune Defic Syndr 2004,37:1111-1124.
6 Friis-Moller N, Reiss P, Sabin CA, Weber R, Monforte A, El-Sadr W, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med 2007,356:1723-1735.
7 Morgello S, Mahboob R, Yakoushina T, Khan S, Hague K. Autopsy findings in a human immunodeficiency virus-infected population over 2 decades: influences of gender, ethnicity, risk factors, and time. Arch Pathol Lab Med 2002,126:182-190.
8Friis-Moller N, Sabin C, Weber R, d‘Arminio Monforte A, El-Sadr W, Reiss P, et al. Combination antiretroviral therapy and the risk of myocardial infarction. N Engl J Med 2003,349:1993-2003.
9 Perronne C. Antiviral hepatitis and antiretroviral drug interactions. J. Hepatol 2006,44:S119-125.
10 Wilby KJ, Greanya ED, Ford JA, Yoshida EM, P cARTovi N. A review of drug interactions with boceprevir and telaprevir: implications for HIV and transplant patients. Ann Hepatol 2012,11:179-185.
11 Vachon ML, Dieterich DT. The HIV/HCV-coinfected patient and new treatment options. Clin Liver Dis 2011,15:585-596.
12 Soriano V, Sherman KE, Rockstroh J, Dieterich D, Back D, Sulkowski M, Peters M. Challenges and opportunities for hepatitis C drug development in HIV-hepatitis C virus-co-infected patients. AIDS 2011,25:2197-2208.
13 Seden K, Back D, Khoo S. New directly acting antivirals for hepatitis C: potential for interaction with antiretrovirals. J Antimicrob Chemother 2010,65:1079-1085.
14 Chan-Tack K, Edozien A. Ritonavir-boosted atazanavir may be efficacious in HIV-infected patients concurrently receiving omeprazole. Clin Infect Dis 2006,42:1344.
15 Eley T, Zhu L, Dragone J, Persson D, Filoromo D, Li T, et al. Effect of omeprazole 20-mg daily on the bioavailability of multiple-dose atazanavir with ritonavir in healthy subjects. (th International Workshop on Clinical Pharmacology of HIV Therapy 2007,April 16-18, 2007, Budapest, Hungary.
16 Furtek K, Crum N, Olson P, Wallace M. Proton pump inhibitor therapy in atazanavir-treated patients: contraindicated? J Acquir Immune Defic Syndr 2006,41:394-396.
17 Kiser J, Lichtenstein K, Anderson P, Fletcher C. Effects of esomeprazole on the pharmacokinetics of atazanavir and fosamprenavir in a patient with human immunodeficiency virus infection. Pharmacotherapy 2006,26:511-514.
18 Department of Health and Human Services. Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection - February 12, 2013. www.aidsinfo.nih.gov 2013,assessed 19 March 2013.
19 Haberl A, Moesch M, Nisius G, Stephan C, Bickel M, Khaykin P, et al. Atazanavir plasma concentrations are impaired in HIV-1-infected adults simultaneously taking a methadone oral solution in a once-daily observed therapy setting. Eur J Clin Pharmacol,66:375-381.
20 AIDSinfo Drug Database: Rilpivirine. http://aidsinfo.nih.gov/drugs/426/rilpivirine/professional 2012,accessed 21.03.2012.
21 Flexner C. The role of plasma protein binding in cART drug-drug interactions. 2013,13th Interna-tional Workshop on the Pharmacology of HIV-Therapy.
22 Celsentri fachinformation. http://www.fachinfo.de/data/fi/ 2011,ViiV Healthcare UK Ltd, Brentford, UK.
23 KKGT. Stichting Kwaliteitsbewaking Klinische Geneesmiddelanalyse en Toxicologie 2014, http://www.kkgt.nl/.
24 INSTAND. INSTAND e.V. Gesellschaft zur Förderung der Qualitätssicherung in medizinischen Laboratorien e.V. 2014, http://www.instandev.de/.
25 Fellay J, Marzolini C, Meaden E, Back D, Buclin T, Chave J, et al. Response to antiretroviral treatment in HIV-1 infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet 2002,359:30-36.
26 Neely M, Decosterd L, Fayet A, Lee JS, Margol A, Kanani M, et al. Pharmacokinetics and pharmacogenomics of once-daily raltegravir and atazanavir in healthy volunteers. Antimicrob Agents Chemother 2010,54:4619-4625.
27 Wenning LA, Petry AS, Kost JT, Jin B, Breidinger SA, DeLepeleire I, et al. Pharmacokinetics of raltegravir in individuals with UGT1A1 polymorphisms. Clin Pharmacol Ther 2009,85:623-627.
28 Burger DM. Raltegravir: a review of its pharmacokinetics, pharmacology and clinical studies. Expert Opin Drug Metab Toxicol 2010,6:1151-1160.
29 Schipani A, Wyen C, Mahungu T, Hendra H, Egan D, Siccardi M, et al. Integration of population pharmacokinetics and pharmacogenetics: an aid to optimal nevirapine dose selection in HIV-infected individuals. J Antimicrob Chemother 2011,66:1332-1339.
30 Wyen C, Hendra H, Siccardi M, Platten M, Jaeger H, Harrer T, et al. Cytochrome P450 2B6 (CYP2B6) and constitutive androstane receptor (CAR) polymorphisms are associated with early discontinuation of efavirenz-containing regimens. J Antimicrob Chemother 2011,66:2092-2098.
31 DAIG. Leitlinien zur Therapie der HIV Infektion in Erwachsenen. 2012, http://www.daignet.de/site-content/hiv-therapie/leitlinien-1.