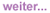47Th Interscience Conference on Antimicrobial Agents and Chemotherapy
(ICAAC),
Chicago, 16.-20. September 2007
Infektiologie auf hohem Niveau
Die ICAAC kann man sicher mit Recht als den "Infektiologen-Kongress" bezeichnen. HIV/AIDS ist dort nur eines von vielen infektiologischen Teilgebieten. Der Kongress bietet daher stets eine ausgezeichnete Möglichkeit, über den Tellerrand des eigenen infektiologischen Wissens zu schauen. Interessant waren insbesondere die Studien zu den neuen Substanzklassen bei HIV und HCV.
Über 12.000 Teilnehmer hatten sich für den 47. ICAAC in Chicago registriert. Für das Kongresszentrum war das kein Problem. Es fand sogar unbemerkt eine weitere Großveranstaltung statt. Thematisch standen die neuen Substanzen im Mittelpunkt.
MOTIVATE: 48-WOCHEN
Vorgestellt wurden die 48-Wochen-Daten der MOTIVATE-Studien 1 und 2 (#H-718a Lalezari J et al.). In dieser Untersuchung wird der Einsatz von Maraviroc bei Patienten mit klinischem Versagen aller drei Standard-Medikamentenklassen (NRTI, NNRTI und PI) geprüft. Die Ergebnisse bestätigten die auf der CROI vorgestellten Daten. Annähernd doppelt so viele Patienten erreichten unter Maraviroc eine Viruslast unter der Nachweisgrenze (<50 Kopien/ml) wie unter Placebo (Abb. 1).
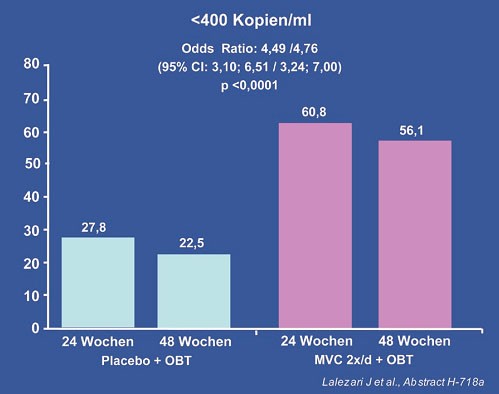
Abb. 1: 48-Wochen-Daten. Maraviroc vs. Placebo bei Drei-Klassenresistenz
Auf der CROI in Los Angeles im Februar 2007 war noch der Eindruck entstanden, die Zugabe von Enfuvirtide zu Maraviroc bringe keinen zusätzlichen Nutzen. Wir wissen aber aus unterschiedlichen Studien bei vorbehandelten Patienten, umso mehr aktive Substanzen, desto besser und länger ist das virologische Ansprechen. Diese Erkenntnis bestätigte sich jetzt auch in der MOTIVATE-Studie, in der die Kombination mit Enfuvirtide eine synergistische Wirkung mit Maraviroc aufwies. Das Nebenwirkungsprofil von Maraviroc war sehr gut. Es gab keinen relevanten Unterschied zu Placebo. Der Trend zu mehr Husten kann bis heute nicht erklärt werden.
TROPISMUS UND VERSAGEN
In einer Subauswertung der MOTIVATE 1 und 2 Studien kam es bei 242/715 (23%) der Patienten in der Maraviroc-Gruppe zum virologischen Versagen (#H-715 Van der Ryst E et al.). Davon hatten beim Versagen 63 ein "dual/mixed tropic" (D/M) bzw. X4-tropes Virus im Vergleich zu 35 Patienten mit R5-tropem Virus. Die Zeit bis zum Versagen war bei den Patienten mit D/M- oder X4-Viren um 30 Tage kürzer. Unter Maraviroc hatten im Vergleich zu Placebo auch die Patienten, die versagten, einen besseren Anstieg der Helferzellen (MVC QD +49 Zellen/µl. MVC BID +71 Zellen/µl vs. Placebo +14 Zellen/µl), wobei dieser Anstieg bei den versagenden Patienten mit CR5-Virus stärker ausgeprägt war. Ob die beobachteten D/M bzw. X4-Populationen bereits als Minorspezies bei Baseline vorlagen und vom Tropismus-Test nicht erkannt wurden, ist unklar.
ARTEMIS: DARUNAVIR/r-FIRSTLINE
Darunavir hat als relativ neuer Proteasehemmer bei mehrfach vorbehandelten Patienten bereits einen festen Stellenwert erreicht. Zum Einsatz bei therapienaiven Patienten wurden die 48-Wochen-Daten der ARTEMIS-Studie vorgestellt (#H-718b DeJesus E et al.). In dieser Untersuchung wurde geboostertes Darunavir 800 mg einmal täglich versus Lopinavir/r an 689 Patienten geprüft. Die Vergleichsgruppe war leider etwas "unsauber". Ein Großteil der Lopinavir/r-Patienten (83%) stellte im Verlauf der Studie von Kapseln auf Tabletten um, 2% nahmen die Kapseln weiter. 77% der Patienten hatten Lopinavir/r zweimal täglich, 15% einmal täglich und die restlichen Patienten wechselten im Verlauf der Studie das Dosierungsintervall. Dieses ist von Bedeutung, da zum einen Lopinavir/r aufgrund des pharmakologischen Profils besser zweimal täglich eingenommen werden sollte und die Lopinavir/r-Kapseln mehr gastrointestinale Nebenwirkungen verursachen als die Tablette. Warum in einer derart aufwändigen Studie solche Unsauberkeiten in der Protokoll-Erstellung auftraten, ist nur schwer nachvollziehbar.
BEI HOHER VIRUSLAST BESSER
Doch nun zu den Ergebnissen. Nach 48 Wochen (die Studie läuft insgesamt 96 Wochen) hatte Darunavir/r das statistische Studienziel der "Nichtunterlegenheit" ("non-inferiority") erreicht. Für den Nachweis einer Überlegenheit ist die Patientenzahl zu klein. Gleich viele Patienten erreichten eine HIV-Plasmavirämie < 50 Kopien/ml (84% vs. 78%) (Abb. 2). Bei hoher Ausgangsviruslast (>100.000 Kopien/ml) war Darunavir/r dem Lopinavir/r statistisch überlegen (DRV 79% vs. LPV 67%, p<0.05) (Abb. 3). In dieser Subgruppe war die Patientenzahl ausreichend groß. Im Hinblick auf Verträglichkeit zeigte Darunavir/r ein gutes Profil und signifikant weniger gastrointestinale Nebenwirkungen (Diarrhöe ab Grad 2: 10% LPV vs. 4% DRV). Die höhere Abbruchrate unter Lopinavir/r kann zumindest teilweise durch die bekannten gastrointestinalen Nebenwirkungen der Lopinavir/r-Kapseln erklärt werden.
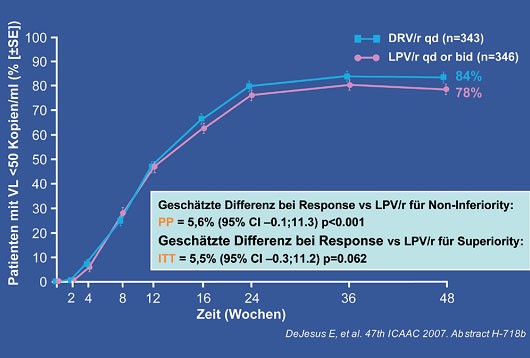
Abb. 2: ARTEMIS: Viruslast <50 Kopien/ml zu Woche 48
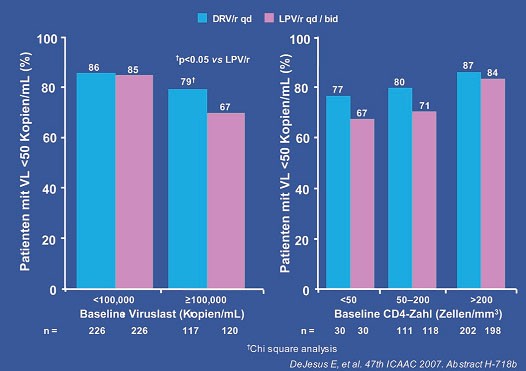
Abb. 3: ARTEMIS: Ansprechen zu Woche 48 in Abhängigkeit von VL oder CD4 bei Baseline (ITT-TLOVR)
RALTEGRAVIR: WEITER "ON THE WAY"
Auf jedem der Kongresse in diesem Jahr gab es neue Daten zu Raltegravir (RAL). Auf der 47. ICAAC wurden die 48-Wochen-Daten der Studie 005 vorgestellt (#H-713 Grinsztejn B et al.). Es handelte sich um eine doppelblinde Dosisfindungsstudie. Dabei erhielten Patienten mit Drei-Klassenresistenz und Therapieversagen 200, 400 und 600 mg Raltegravir BID plus eine optimierte Begleittherapie (OBT) nach Resistenztestung. Die Ergebnisse bestätigen das bisherige sehr gute Ansprechen selbst bei Multiresistenz. Zu Woche 48 hatten 64-71% der Patienten eine Viruslast <400 Kopien/ml, 46-64% sogar <50 Kopien/ml (Abb. 4).
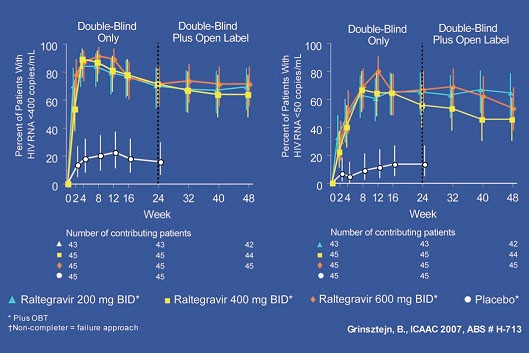
Abb. 4: Raltegravir 200, 400 und 600 mg BID vs. Placebo. Nachdem die Standarddosis von Raltegravir auf 400
mg BID festgelegt worden war, konnten nach 24 Wochen alle Patienten aus den verblindeten Dosierungsarmen
auf diese Dosierung umgestellt werden.
RALTEGRAVIR-NEBENWIRKUNGSPROFIL
Das Nebenwirkungsprofil von Raltegravir sah sehr gut aus. Auch in dieser Studie war es wieder mit Placebo vergleichbar. Die meisten Nebenwirkungen waren mild bis moderat, wobei die Laborabweichungen leider wie so oft erst ab Grad 3 gemeldet wurden. Eine Erhöhung der GOT und GPT beispielsweise wird bis zu einer Erhöhung zum 5fachen der Norm als AE Grad II gewertet und wird somit nicht in der Präsentation gezeigt. Das betrifft Erhöhungen bis 250 IU/l . Bei einem Patienten unter RAL musste wegen Erhöhung der Leberwerte die Therapie abgebrochen werden. Des Weiteren traten vier schwere unerwartete Nebenwirkungen auf. Unter Raltegravir wurde eine akute Pankreatitis nach der zweiten Dosis sowie ein Todesfall bei metabolischen Azidose, Nierenversagen und Sepsis beobachtet, unter Placebo ein Schlaganfall und eine Verschlechterung einer Lipoatrophie.
RALTEGRAVIR-RESISTENZEN

Fotos:Thomas Buhk, Hamburg
Resistenzen werden unter Therapieversagen mit Raltegravir häufig beobachtet. Dieses spricht bei dieser hochaktiven Substanz für eine eher niedrige Resistenzbarriere. In der vorgestellten Studie fand sich ein virologisches Versagen bei 38/133 (29%) Raltegravir-Patienten. 68% davon hatten einen GSS von 0, d.h. keine weitere genotypisch wirksame Substanz in der Begleittherapie.
Genotypische Daten liegen für alle 38 Patienten vor. Die meisten wiesen Integrase-Mutationen auf (35/38). Bei 34/35 konnten zwei genetische "pathways" nachgewiesen werden: N155 oder Q148. Die Resistenz war typischerweise mit zwei oder mehr Mutationen assoziiert (31/35 Patienten), bei 13 Patienten mit Q148H und/oder G140S. Es bestand keine Korrelation zwischen Resistenzentwicklung und Medikamentkonzentration.
INTERLEUKIN-2: SPÄTER ZUR HAART?
Aus Frankreich hören wir regelmäßig seit Jahren Nachrichten über den möglichen Einsatz von Interleukin 2 (IL-2). Die jetzt vorgestellte Arbeit (#H-718 Molina J et al.) untersucht den Einfluss von Interleukin auf den Zeitpunkt des Therapiebeginns bei asymptomatischen Patienten. ART-naive Patienten (CD4 300-500/µl) wurden entweder beobachtet (n=64) oder mit IL-2 behandelt (n=66, 4.5 Mio s.c. BID für 5 Tage, dann weitere Zyklen an Woche 8, 16, 24 und zwei Zyklen zwischen Woche 48 und 80). Endpunkte waren CD4-Zellen <300/µl, ART-Beginn oder eine AIDS- definierende Erkrankung oder Tod.
Zu Woche 96 kam es bei 35% der IL-2 Patienten verglichen mit 59% im Kontrollarm zum Auftreten eines Endpunktes (p=0.005). Die CD4-Zellen stiegen unter IL-2 um +51/µl an, im Kontrollarm fielen sie um -64/µl (p=<0.0001).
Der Beginn der ART konnte bei den IL-2 Patienten um 48 Wochen verschoben werden (p=0.02).
WENIGER ZERVIX-KARZINOME UNTER HAART?
HIV-positive Frauen haben ein höheres Risiko für die Entwicklung eines Zervix-Karzinoms. Ob die HAART dieses Risiko vermindert, ist bisher nicht bekannt. Eine Gruppe aus Barcelona (#H-1727 Clotet B at al.) ging dieser Frage in einer retrospektiven Analyse nach.
Eingeschlossen wurden Frauen mit zwei normalen Zervix-Abstrichen seit 1997 sowie CD4-Zellen >350/µl. Intraepitheliale Läsionen (SIL) im Abstrich fanden sich bei 30% der Frauen unter HAART (n=90) und 19% der Frauen ohne HAART (n=37). Die Wahrscheinlichkeit, keine SIL innerhalb von drei Jahren zu bekommen, war vergleichbar (70% vs. 78%).
HEPATITIS C: PROTEASE-INHIBITOREN
Der Proteaseinhibitor Telaprevir wird vom Unternehmen Vertex hergestellt und in Kooperation mit Tibotec entwickelt. In der Phase-II-Studie PROVE-1 wird die neue Substanz an Patienten mit HCV-Monoinfektion und Genotyp 1 geprüft (#V-1383, Sulkowski M.S. et al.). Die Studie hat vier Gruppen. Die Kontrollgruppe erhielt die Standardtherapie Peg-Interferon-2a (pegIFN) und Ribavirin (RBV) über 48 Wochen (n=81) sowie drei weitere Gruppen mit TVR 750 mg TID, pegIFN 180 µg/W und RBV 1.000-1.200 mg/d für 12 Wochen gefolgt von O Wochen (n=20), 12 Wochen (n=80) und 36 Wochen (n=82) Therapie. Patienten mit negativer HCV-Virämie zu Woche 4 haben erfahrungsgemäß eine deutlich höhere Chance zum Therapieerfolg.
Eine solche RVR (Rapid Virologic Response) wurde bei 11% der Patienten unter Standardtherapie beobachtet, jedoch bei bis zu 79% der Patienten in den drei Telaprevir-Armen (p<0.001). Bei Woche 12 waren 39% (Kontrolle) der Patienten vs. 70% (TVR) virusfrei (p<0.001).
INTERAKTION: ABACAVIR - RIBAVIRIN
Beide Substanzen sind Guanosin-Analoga, eine intrazelluläre Konkurrenz um die Phosphorylierung ist somit denkbar. Die spanische Arbeitsgruppe um Soriano hat sich dieser Frage in einer retrospektiven Analyse angenommen (#H-1731 Vispo E et al.). Es wurden die Daten von 426 HIV/ HCV-Koinfizierten (80% Männer, 41 Jahre, CD4 567/µl) ausgewertet. Alle waren immunologisch stabil. Die HCV-RNA betrug 5,8 logIU/ml, 65% hatten Genotyp 1 oder 4. Alle wurden mit einer Kombination von pegIFN und RBV behandelt. Insgesamt kam es bei 72% zum HCV-Therapieversagen. In statistischen Auswertungen waren verschiedene Faktoren hierfür verantwortlich, eine hohe HCV-Virämie, Genotyp Grad 1 und 4, niedrige RBV-Plasmaspiegel und der Einsatz von Zidovudin oder Abacavir. Der negative Effekt von Abacavir war nur in der Subpopulation der Patienten mit tiefem Ribavirin-Plasmaspiegel nachweisbar (<2 µg/ml).
HEPATITIS B: HBsAg-MUTATIONEN
Der serologische Nachweis von HBV basiert auf dem Nachweis des "small surface" Antigens (HBsAg). Punktmutationen könnten theoretisch die kommerziell verfügbaren Teste beeinflussen. Eine französische Arbeitsgruppe untersuchte die Prävalenz des HBsAg Polymorphismus bei insgesamt 169 chronisch- oder neuinfizierten Patienten (#V-1903 Roque-Afonso SR et al.). 27,3% der Patienten hatten in der MHR-Region genetische Varianten. Bei den chronisch infizierten Patienten waren es sogar 43,1%. Das Auftreten der MHR-Varianten war signifikant assoziiert mit Alter und Lamivudin-Resistenz.
Dr. Olaf Degen
Universitätsklinikum Hamburg-Eppendorf
Ambulanzzentrums GmbH · Bereich Infektiologie
Martinistr. 52 · 20246 Hamburg
E-Mail: degen@uke.uni-hamburg.de